The basics of molecular dynamics starts from a simple fact from thermodynamics: the most probable thermodynamic state is the state which has the lowest thermodynamic free energy. Thus, the goal of molecular dynamics is to study the statistical properties of possible molecular configurations, which is completely described by the Partition Function.
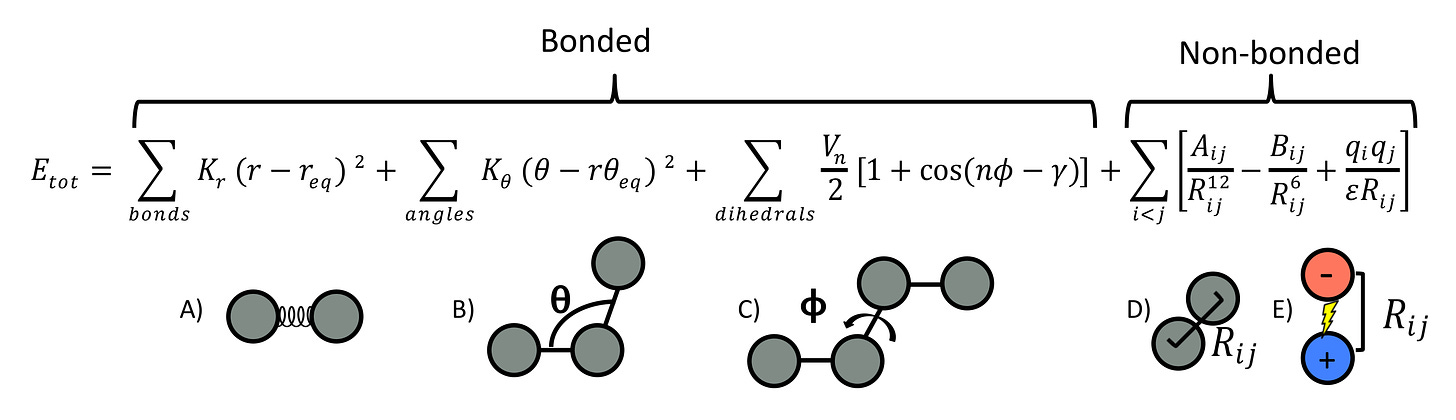
The molecular structure with the lowest free energy is determined by the forces acting on the molecule. Generally, there are 4 forces – bond forces , electrostatic forces, Van der Waals forces, and solvent interactions (e.g. hydrophobic interactions). However, a computer cannot perfectly represent all 4 forces. Therefore, some assumptions about the force field are necessary (e.g. ignoring the flexibility of water molecules, or ignoring quantum effects).
Depending on the type of assumptions, there are multiple force field approximations, such as CHARMM, AMBER, or GROMACS. However, there are some common features. Generally, the force field is taken as the negative gradient of the potential, that is
This differential equation is solved by means of time-integration, where time is divided into discrete steps with a order of approximately 1 femto-second.
The time-evolution of the
Meanwhile,
This can be calculated as